Version 2024 of NMR Predictors adds new features, including the ability to predict spectra of multiple structures in separate windows, and updated prediction databases. Read below for details, and contact us for help upgrading your software.
Data Import
- You can now import 1H and 13C NMR data, structures, and assignments from third-party generated JCAMP-DX format files into the prediction database
- You can now import data from the following formats:
- MS data from Bruker TIMS TOF (*.tsf)
- GC data from Shimadzu (*.gcd)
- We improved data import from:
- Standard BMRB bioNMR (*.seq and *.bmrb) biosequence structure and assignment data is now transferred to C-H correlation spectra (HSQC and HMQC), in addition to the previous N-H HSQC
Predict Spectra of Multiple Structures in Separate Windows
- You now have the option to predict individual spectra of a particular nucleus or nuclei for multiple structures at once, in addition to predicting them as components of a mixture or as a single structure with continuous numbering.
Option to Define the 1H Range of Multiplets Based on Peaks
- You now have the option to report the chemical shift range of multiplets based on the outermost peaks in the Multiplet Report Options dialog box, in addition to the boundaries of integration
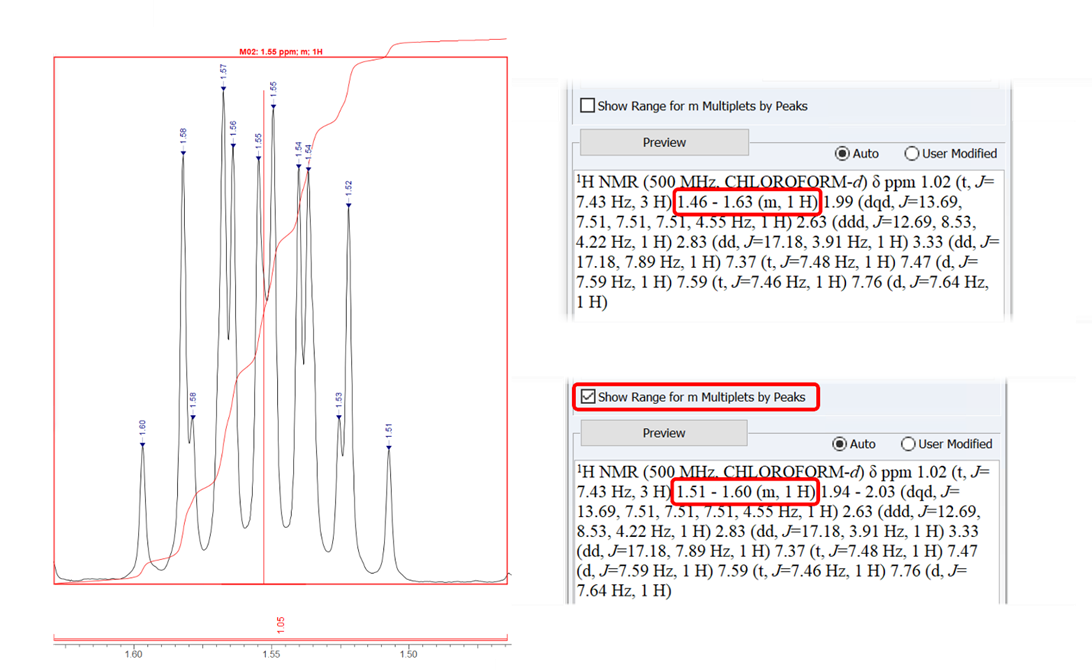 Choose to report multiplet ranges according to the outermost peaks or the boundaries of the integration—either 1.51–1.60 (m, 1H) for the outermost peaks with the option checked (above), or 1.46–1.63 (m, 1H) according to integration boundary (below).
Choose to report multiplet ranges according to the outermost peaks or the boundaries of the integration—either 1.51–1.60 (m, 1H) for the outermost peaks with the option checked (above), or 1.46–1.63 (m, 1H) according to integration boundary (below).
Display Annotations of 1D Curves in 2D Spectra
- You now have the option to display the annotations of 1D curves in 2D NMR spectra
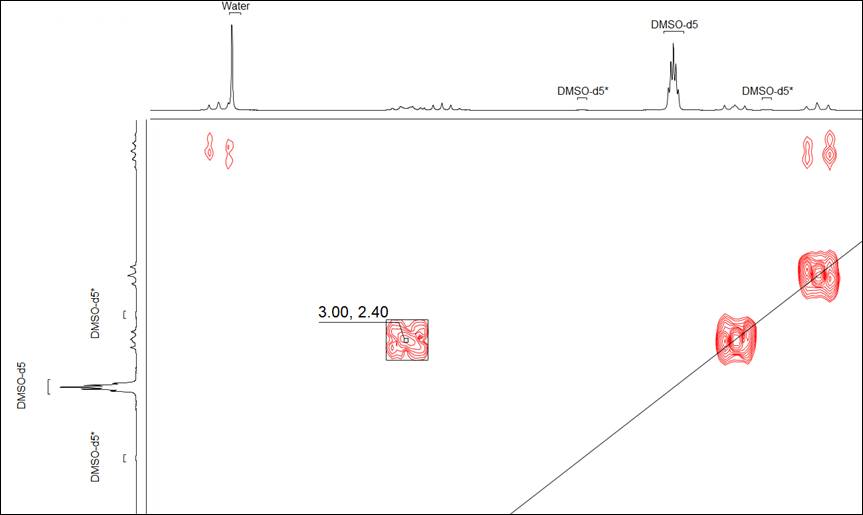
Show or hide the annotations of 1D curves in 2D NMR spectra.
Indication of Peaks Used for qNMR Calculation in Report
- You can now quickly identify which integrals are used in qNMR calculations with external standards in the Table of Integrals; which can now be included on the title page of the report
Option to Automatically Unfold HSQC Spectra for Structures with Si-CH3 Groups
- You now have the option to automatically unfold HSQC spectra along the F1 (13C) axis for structures with Si-CH3 This allows aliased peaks that are cut off to be picked properly.
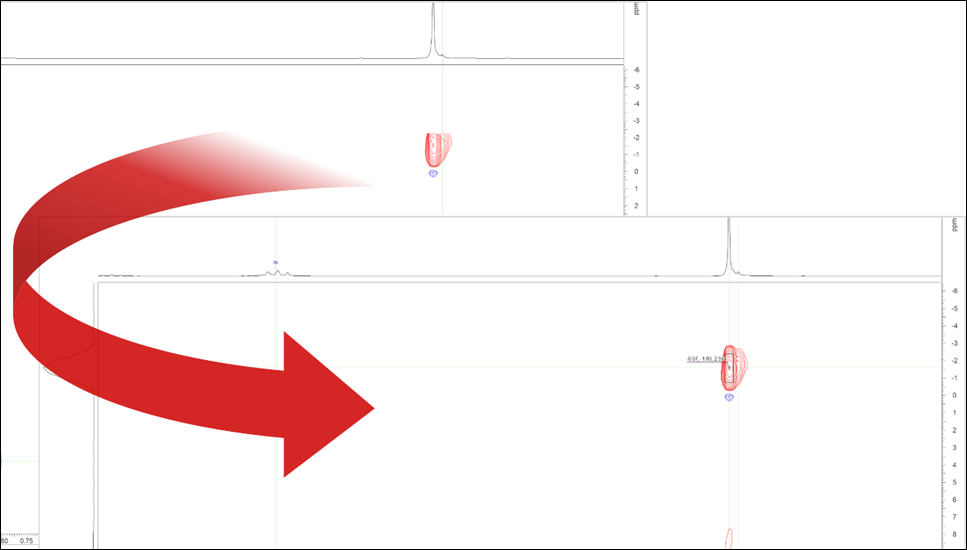
Automatically unfold HSQC spectra along the F1 for structures with Si-CH3 groups.
Updated Prediction Databases
- You can now predict spectra based on databases with thousands more structures, chemical shifts, and coupling constants than ever before
| Structures (difference from previous update) | Chemical Shifts (difference from previous update) | Coupling Constants (difference from previous update) | |
| 13C + 1H NMR Database | 392,075 (+8,095) | 5,609,600 (+219,550) | 1,152,584 (+53,401) |
| 13C NMR Database | 262,052 (+7,580) | 3,741,520 (+154,172) | 152,558 (+9,153) |
| 1H NMR Database | 283,215 (+7,969) | 2,768,019 (+123,582) | 1,044,497 (+44,975) |
| 19F NMR Database | 36,038 (+4101) | 62,402 (+5,429) | 89,264 (+12,996) |
| 15N NMR Database | 10,362 (+34) | 24,657 (+97) | 5,087 |
| 31P NMR Database | 33,216 (+782) | 40,250 (+836) | 51,484 (+2,108) |
New Integral Normalization Method for NMR Spectra
- You can now more accurately normalize 1H integrals of samples with various impurities with the Allow Excess option
- This can help prevent false negatives in workflows like Automated Structure Verification (ASV)

The Allow Excess option method for 1H integral normalization can avoid under-estimated integrals when impurities are present in the sample, which helps prevent false negative results in ASV.
- This can help prevent false negatives in workflows like Automated Structure Verification (ASV)
Perform External Standard Concentration Calculations Without a Sample Structure
- You now have the option to perform qNMR external concentration calculations in cases where the structure of the sample is not available. Do this by entering the molecular weight of the sample in the Concentration dialog box.
Improved Metrics for External Standard qNMR
- You can now calculate the relative sample purity by weight (w/w%) automatically with the Concentration Calculation tool
- You can now include the concentration calibration parameters in your report
Option to Show Only Unique J-Coupling Constants in the NMR Spectral Data Table
- You now have the option to show only unique coupling constants in the Spectral Data Table Advanced Options menu
- Instead of reporting a triplet as t(5.25, 5.25), it can now be reported as t(5.25)
Improvements to Search Capabilities
Monoisotopic Mass Included in Table of Search Results by Default
- You can now automatically see the ‘Monoisotopic Mass’ column in the Table of Search Results
New Compensated Offset Spectrum Similarity Search Method for NMR
- You can now better search for spectra where the query and the hit have very similar multiplet structures, but the peaks are shifted by different amounts. This shift is often seen between spectra recorded at different pHs or temperatures, and can now be accounted for with the new Compensated Offset method of HQI calculation

New Approximate Mode for 2D NMR Spectral Search
- You can now search for 2D NMR spectra without exact peak matches with Approximate Mode
- Similar to 1D spectra, this search mode ignores the number of peaks existing in the search zones in the query and hits. The HQI is then defined by the ratio of corresponding search zones in the spectra.
As always, you can process data from other analytical techniques in NMR Predictors. We’ve improved features for these techniques as well:
Processing Hyphenated MS Data with NMR Predictors
Improved Display and Reporting of Structures on Chromatographic Traces
- You now have the option to selectively display structures on chromatographic traces, consolidating information in one place. This feature allows you to control which structures to display and include in reports.
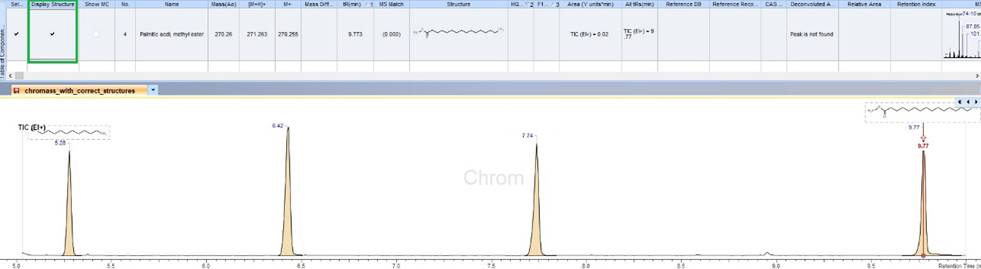
Display selected structure(s) on chromatographic traces.
Simplified Analysis with Reoccurring Ions
- You can now add reoccurring ions to the Component Interpretation Options, allowing you to automatically search and label reoccurring ions when assigning a component
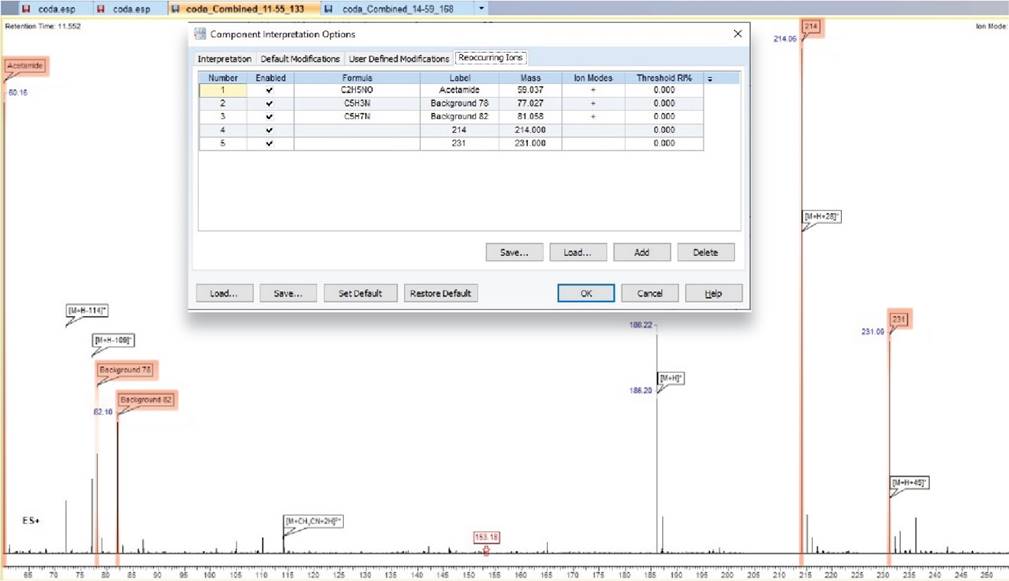
Highlighted ions are those populated in the reoccurring ions tab and are clearly labeled on the spectrum for ease of interpretation.
Greater Confidence in Identification with Improved MS Match Score
- You can now include multiply-charged ions and/or modifications into your MS Match score, giving you greater confidence in identification
Improvements in the Use of Internal Standards in Hyphenated Data Sets (LC/UV/MS, GC/MS)
- You can now calculate relative areas for all components if one component is selected as the internal standard
Easier Export of Components to Excel
- You now have the option to selectively push results including structure and molecular formulae from the Table of Components to Excel
Improved Accuracy in Area Calculations
- Using improved peak picking for XICs you can now manually adjust the border and/or baseline of peaks on XIC traces
Improved Efficiency of MS Data Processing
- You can now use preset options for Profiles to link instruments to specific accuracy, reporting, preferences, and script settings
- Set known parameters by instrument to minimize errors and improve efficiency
Improved Visualization of MS and UV Spectra
- You can now more easily review data with fixed positions for UV and MS Spectra
- Component Spectra are permanently fixed on the left and scan spectra are on the right
Improved Regulatory Compliance
You can easily store and visualize XIC accuracies and identify the accuracy used to extract XICs.
- Optionally display accuracies in the LC/UV/MS legend, in the chromatogram legend, and appended to the chromatogram name
- Visualize any accuracy changes in the History Record
- Store and display XIC accuracy in Spectrus DB, and visualize in reports
Improved Reporting Functionality
We have added more options for reporting functionality including:
- Ability to include full and deconvoluted areas in reports
- Ability to zoom to a defined region around an MSn Precursor
- Report “All” wavelengths for flat UV traces inside hyphenated datasets
- Display multiple descriptors on peaks in flat chromatograms such as retention time and name
Ease of Use Improvements
- You can access all available options in mass spectral right-click menus at once
- Quickly restore your zoom with a new hotkey for zoom undo (“U”)
Want to learn more?
Read more about the full features of NMR Predictors, or contact us for help upgrading your software.