Consolidate all your process chemistry data and avoid data paralysis with Luminata®! Find out more.
Current Software Versions
What's New in PhysChem Suite™ Version 2024
Version 2024 of PhysChem Suite introduces dramatic enhancements in the prediction accuracy for pKa, logP, and logD, as well as related pH profile of aqueous solubility and ADME endpoints such as absorption, bioavailability, and blood-brain barrier penetration, as a result of thorough algorithm improvement paired with significant expansion of the training sets. Read below for details, and contact us for help upgrading your software.
Significant revisions and improvements to the pKa, logP, and logD algorithms and equations with particular focus on complex heterocycles and novel therapeutic modalities (PROTACs)
Further significant enhancements to the pKa Classic algorithm, building upon the previously released improvements, including the addition of more than 5000 experimental pKa values from diverse chemical space, including aromatic and aliphatic heterocycles
LogP GALAS training set of experimental values has been increased by 25% (~4750 new experimental logP values) after careful curation, reinterpretation, and verification of new data. This adds exciting accuracy improvements for the varied and diverse chemical classes.
Technical improvements to the logD algorithm, in addition to enhanced accuracy prediction as a result of improved logP and pKa predictors
You can expect greatly improved accuracy of pKa calculations due to a significant expansion of the algorithm training sets, with several diverse chemical datasets. We have developed novel algorithms and deeply revisited models for specific classes. Data quality and chemical diversity guide our training efforts for each release. Through our own research, and ongoing partnership initiatives, our algorithm training expands covered chemical space, and allows us to enrich, curate, and prioritize historically available training data, for better predictive performance.
After the addition of new datasets, the full ACD/Labs pKa training data set presently contains ~26,000 compounds with more than 42,000 experimental pKa values. It should be noted that not all compounds and pKa values are always used for algorithmic Hammet equations or pKa0 values. However, with new datasets available, previously unused “old” data can be used for new algorithm improvements.
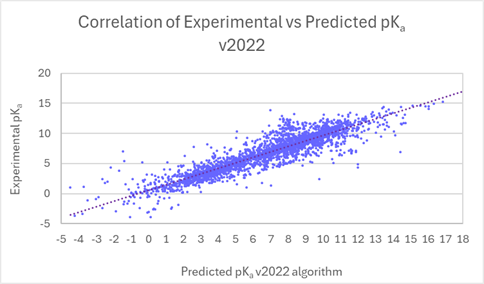
The improvements in the pKa prediction accuracy using the historically renown BioByte Master file from v2022 to v2024 are shown in the table below:
Number of predicted pKa values
Average prediction accuracy
v2022
12,224
0.419
v2023
12,421
0.412
v2024
12,458
0.395
Improved pKa Prediction Accuracy for Expanded Pharmaceutical Space
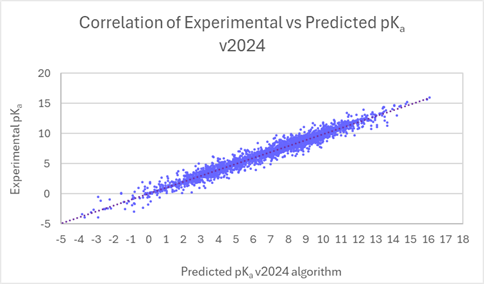
You can have greater trust in the accuracy and reliability of pKa calculations from a significant expansion of the pKa Classic algorithm training set. A brand-name pharmaceutical company has provided data for over 1100 compounds with more than 2200 pKa values for this project. The dataset contained exciting modern compounds and members of chemical classes underrepresented in previous versions of our software. Collaborative efforts allowed us to improve prediction accuracy for this chemistry, which has resulted in overall improvement of pKa predictions for current and future users of the software. The linear scatter plots below present the comparison of experimental versus predicted pKa, before and after training (v2023 versus v2024) for the new data set provided by the pharmaceutical company. The average error of prediction is significantly lower in v2024 (0.27 log units vs. 0.76 log units in v2023, and R2 is closer to 1 (0.98 vs. 0.86 in v2023).
Linear regression plots demonstrate the improved correlation between experimental and predicted pKa (pKa Classic algorithm) for 2200 ionization centers from v2023 to v2024.
Training of the algorithm resulted in significant improvements in prediction accuracy for these 1100 novel pharmaceutical compounds, with more ionization centres calculated using v2024 (2262 vs. 2248 in v2023). 84% of pKa values for these compounds are now predicted within 0.5 log units (99% within 1 log unit). This compares to 47% within 0.5 log units (72% within 1 log unit) in v2023.
Improvement in prediction accuracy (pKa Classic algorithm) for 2200 ionization centers in 1100 pharmaceutical compounds in v2024.
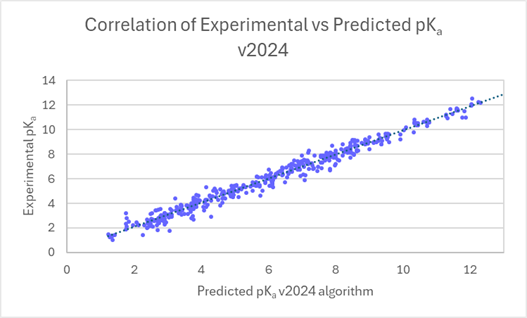
Enhanced pKa Prediction Accuracy for Diverse Chemical Compounds
You can now expect significant improvement in the prediction accuracy of pKa for compounds belonging to diverse chemical classes, such as aromatic and aliphatic heterocycles which were previously underrepresented in our training sets. Working with publicly available data sources, and a government organization, we have augmented a training dataset using opensource data with 3150 curated ionization centers from 2500 chemical compounds.
The linear scatter plots below show the comparison of experimental vs. predicted pKa, before and after training. The average error of prediction is significantly lower in v2024 (0.43 log units vs. 0.61 log units in v2023 and 0.88 in v2022), and R2 is closer to 1 (0.98 versus 0.93 in v2023 and 0.83 in v2022).
Training of the algorithm resulted in significant improvements in prediction accuracy for these 2500 compounds, with more ionization centres calculated using v2024 (3158 vs. 3023 in v2023). 68% of pKa values for these compounds are now predicted within 0.5 log units (91% within 1 log unit). This compares to 58% within 0.5 log units (81% within 1 log unit) in v2023, and to 48% within 0.5 log units (70% within 1 log unit) in v2022.
Improvement in prediction accuracy for 3150 ionization centers in 2500 compounds in v2024.
Enhanced Prediction for Novel Therapeutic Modalities of High Interest
You can now expect improved pKa predictions for PROTACs (Proteolysis Targeting Chimeric Molecules). We have collected data on PROTACs and their precursors with measured experimental pKa values that originated from both our industry collaborators and public sources.
PROTACs are composite molecules designed to attenuate function of specific proteins by binding to the target protein and inducing its degradation by intracellular ubiquitin-proteasome system. Structurally, PROTACs are heterobifunctional molecules, composed of two small molecular ligands connected by a covalent linker (one ligand is responsible for recognition of the target protein, another for the recruitment of ubiquitin E3 ligase, initiating the degradation pathway). Overall, this results in larger molecules that fall beyond the traditional Rule-of-five governed chemical space and therefore may prove challenging for various property prediction algorithms.
Two comparisons below present the results of the enhanced prediction between v2023 and v2024.
The linear scatter plots below show the comparison of experimental versus predicted pKa of this data, for version 2023 and the new version 2024. The average error of prediction is lower in v2024 (0.30 log units vs. 0.42 log units in v2023).
Linear regression plots demonstrate the improved correlation between experimental and predicted pKa values for 49 ionization centres of PROTACs reviewed in the publication
Examples of PROTACs and the comparable predicted pKa values from v2024
Exp. pKa
Pred. pKa v2024
Exp. pKa
Pred. pKa v2024
2.74
2.90
4.69
3.85
6.27
6.02
7.98
7.82
Overall PROTAC Assessment
The linear scatter plots below show the comparison of experimental vs. predicted pKa, for v2023 and v2024 for a dataset of 253 PROTAC molecules with 491 ionization centers. The average error of prediction is significantly lower in v2024 (0.28 log units vs. 0.52 log units in v2023).
Linear regression plots demonstrate the improved correlation between experimental and predicted pKa for 491 ionization centers from v2023 to v2024.
Improved Accuracy of the LogP GALAS and Resulting LogP Consensus Algorithms
You can have greater trust in the calculations from a significant expansion of the logP GALAS algorithm training set. For the most part, the newly collected data represents logD7.4 values that have been back-calculated to logP using the ACD/pKa Classic algorithm (which features improvements to minimize uncertainty related to evaluation of the compound’s ionization state).
Improvements include:
Inclusion of ~4750 new experimental values
Increase in the number of entries to more than 22,000 (>25% increase in entries overall)
Addition of series of compounds with novel scaffolds and varying substituents, such as heterocycles and functional groups which are usually seen as challenging to source and predict
Improved prediction accuracy of high MW compounds, such as PROTACs
The improvements in calculation accuracy are available with both ACD/LogP GALAS and ACD/LogP Consensus algorithms utilizing the new LogP v. 1.4 built-in self-training library.
The linear regression scatter plots below show the comparison of experimental vs. predicted logP values for newly acquired compounds, before and after the training.
Linear regression plots demonstrate the improved correlation between experimental and predicted logP values for ~4750 compounds from v2023 to v2024.
Examples of PROTACs and the comparable predicted logP values from v2024 and v2023.
Exp. LogP
Pred. v2023
Pred. v2024
Exp. LogP
Pred. v2023
Pred. v2024
4.23
5.68
4.44
4.21
2.81
3.87
Reliability index
0.44 (Borderline)
0.86
(High)
Reliability Index
0.49 (Borderline)
0.75
(High)
Predict LogD More Accurately
You can now expect greater accuracy for these calculations due to several technical improvements to the algorithm, including enhancements to the normalization procedure
Inaccuracies between the simulated logD pH curves and the plot expected from the compound’s apparent pKa profile have been resolved
Starting from the current release, the shape of the simulated logD pH curve will closely match the shape expected from the compound’s apparent pKa values
You can have greater trust in the accuracy of logD predictions and water solubility pH profile due to improvements in the pKa and logP algorithms
Updated Reporting Templates
You can generate compliance reports with confidence as the QPRF report templates have been updated to v2.0 in full accordance with QAF checklist.
Improved Batch Reporting
You can now automatically save all individual reports as a .zip file, preventing duplication of file names
Improved Export of Data
With the latest update you can now:
Export your data in the less limiting .xlsx file format
Export molecular structures as vector images which offers better scaling support
Include atom numbers with structure images, enabling you to easily analyze atom-specific properties
Convert Units in Calculations
You can now easily configure the software to convert certain units of calculated values to user-specific dimensions by default (for example converting concentration from molar to mass)
ACD/Labs’ development team is eager to collaborate with organizations to improve predictions for novel compounds. Do you have accurately measured experimental values for the predictions we support? Contact us to discuss how we may work together.
To provide the best experiences, we use technologies like cookies to store and/or access device information. Consenting to these technologies will allow us to process data such as browsing behavior or unique IDs on this site. Not consenting or withdrawing consent, may adversely affect certain features and functions.
Functional
Always active
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
Preferences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistics
The technical storage or access that is used exclusively for statistical purposes.The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
Marketing
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.
To provide the best experiences, we use technologies like cookies to store and/or access device information. Consenting to these technologies will allow us to process data such as browsing behavior or unique IDs on this site. Not consenting or withdrawing consent, may adversely affect certain features and functions.
Functional
Always active
The technical storage or access is strictly necessary for the legitimate purpose of enabling the use of a specific service explicitly requested by the subscriber or user, or for the sole purpose of carrying out the transmission of a communication over an electronic communications network.
Preferences
The technical storage or access is necessary for the legitimate purpose of storing preferences that are not requested by the subscriber or user.
Statistics
The technical storage or access that is used exclusively for statistical purposes.The technical storage or access that is used exclusively for anonymous statistical purposes. Without a subpoena, voluntary compliance on the part of your Internet Service Provider, or additional records from a third party, information stored or retrieved for this purpose alone cannot usually be used to identify you.
Marketing
The technical storage or access is required to create user profiles to send advertising, or to track the user on a website or across several websites for similar marketing purposes.