Version 2023 of Method Selection Suite improves method transfer between instruments and labs, among other features. Read below for details, and contact us for help upgrading your software.
Improved Method Transfer Between Instruments and Labs
You now have new options to improve method transfer between instruments and labs, and simplify your workflow including:
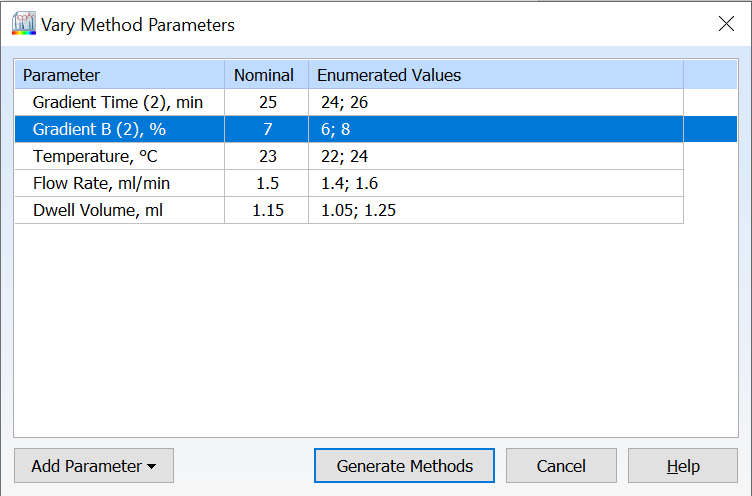
- All available method parameters are shown by default in the Vary Parameters window
- Varied parameters are highlighted for a quick visual of what has been modified with the option to reset all parameters
- Easily view the number of methods to be generated before proceeding with your varied parameters
- Retain or remove your last used methods if you choose to vary parameters more than once
The below features were included in the software in Spring 2023.
Transfer Methods without Impacting Method Performance by Varying Optimization Parameters
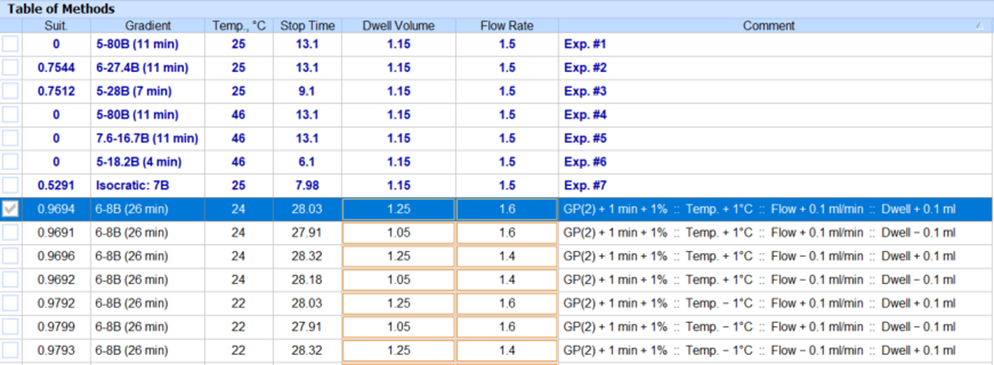
- You can now vary column optimization parameters and autogenerate methods to aid method transfer between groups or CxO partners without impacting method performance.
- Modify column optimization parameters for methods (e.g., flow rate, column length, column diameter, dwell volume, etc.)
-
- Autogenerate a new group of methods with all possible combinations of varied parameters to ensure coverage of relevant method space.
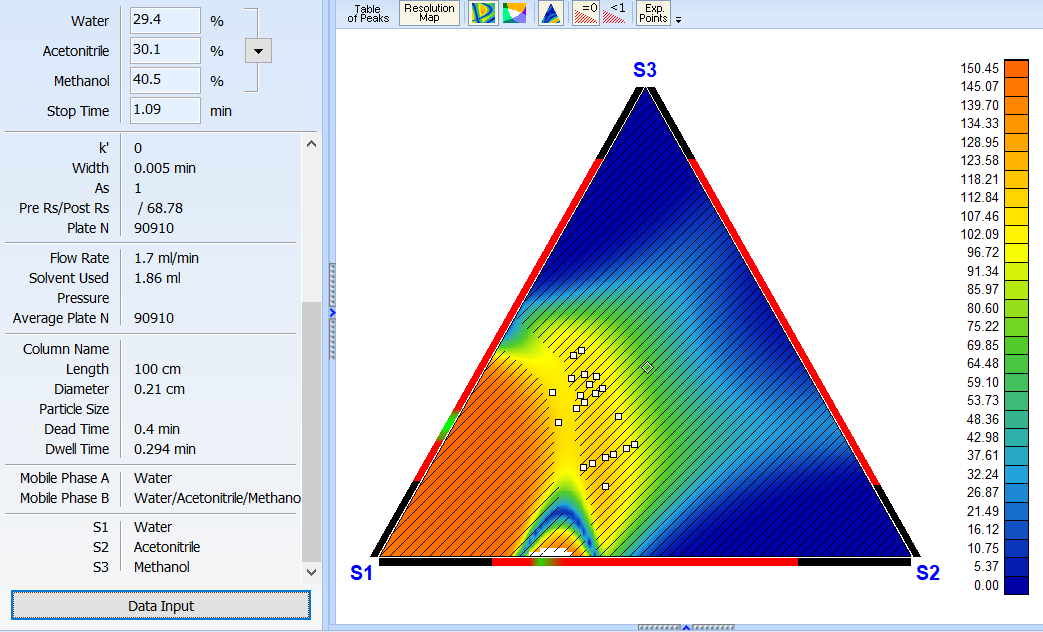
Improved 2D Solvent Ratio Optimization Mode
For cases where three primary organic phases cannot be determined within the provided experiments, the software will try to use the pure solvent components from the given experimental mobile phases (S1, S2, S3) as vertices of the optimization region.
Predict pKa More Accurately
You can have greater confidence in calculations from a significant expansion of the pKa Classic algorithm training set. Improvements comprise:
- Inclusion of ~4000 environmental, pharmaceutical, and chemical compounds to the training set
- An increase in the number of experimental pKa values to more than 40,000
- Rebuilding of existing Hammet-type and new equations for novel chemical classes
- Addition of new ionization centers
Improved Accuracy of LogD
Improvements to the pKa Classic algorithm directly improve the accuracy in pH and logD calculations.
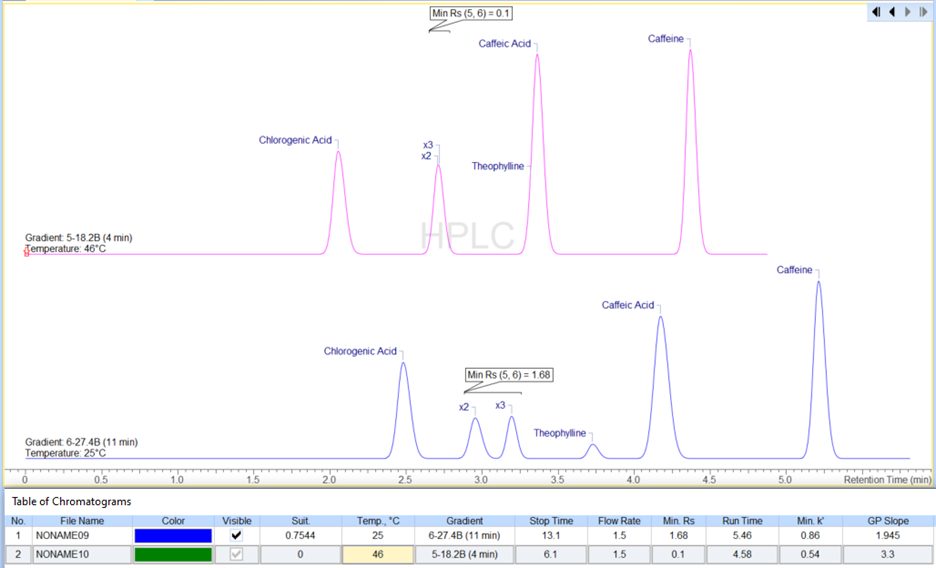
Better Visualization of Chromatograms
You now have easier access to use the Adaptive Time Axis to zoom in on your peaks of interest.
Improved Data Input Window
Quickly remove experiments or copy/paste method parameters between tabs from simulations.
Ease of Use
Set method details (i.e., gradient, flow rate, etc.,) once and apply across all traces
The below features were included in the software in Spring 2023.
Select and Transfer Multiple Methods Simultaneously
- New columns containing method data (e.g., critical pairs, start %B duration, etc.) have been added.
- You can now select multiple methods for deletion or transfer to the processing interface.
- Selected simulated chromatograms can be transferred to the processing interface as individual datasets or series (with relevant metadata e.g., critical pairs, gradient program, run time, Min Rs, etc.) Previously, you could only select and transfer one method at a time.
- We improved data import from:
- Agilent OpenLab ChemStation and Bruker LC/MS ToF DAD traces—DAD data and extracted wavelengths can now be imported and viewed
- We now support Shimadzu LabSolutions CDS data from LCMS-9050 Q-TOFs
- We now support import from archives (i.e., *.zip)
- Thermo Scientific Chromeleon CDS add-on is improved to display m/z values for SIM data
- OpenLab CDS add-on now uses a single processing method to export all datasets in the OpenLab CDS repository
- We have expanded the data you can export to JSON format for use in data science and modeling
- Export hyphenated xC/UV/MS data
- Export processed 1D and 2D NMR spectra, including peaks, multiplets, and assignments
The below features were included in the software in Spring 2023.
- We improved data import from:
- Waters UNIFI and Waters Connect with ACD/Labs “Connect to” tool. You can now import processed results and import and view each file within a project. Import RDa data too.
- OpenLab CDS Add-On—we have increased the peak fields and custom parameters we import
- Xcalibur—you can view the UV wavelength on import and in the data processing interface
- Shimadzu LabSolutions CDS Add-On—we have increased the metadata and peak fields we import
- Bruker CompassXtract—we have accelerated the translation process allowing you quicker access to your data
- Import from Archive (i.e., *.zip) – you can save time by skipping the unzip step for supported archived folder formats
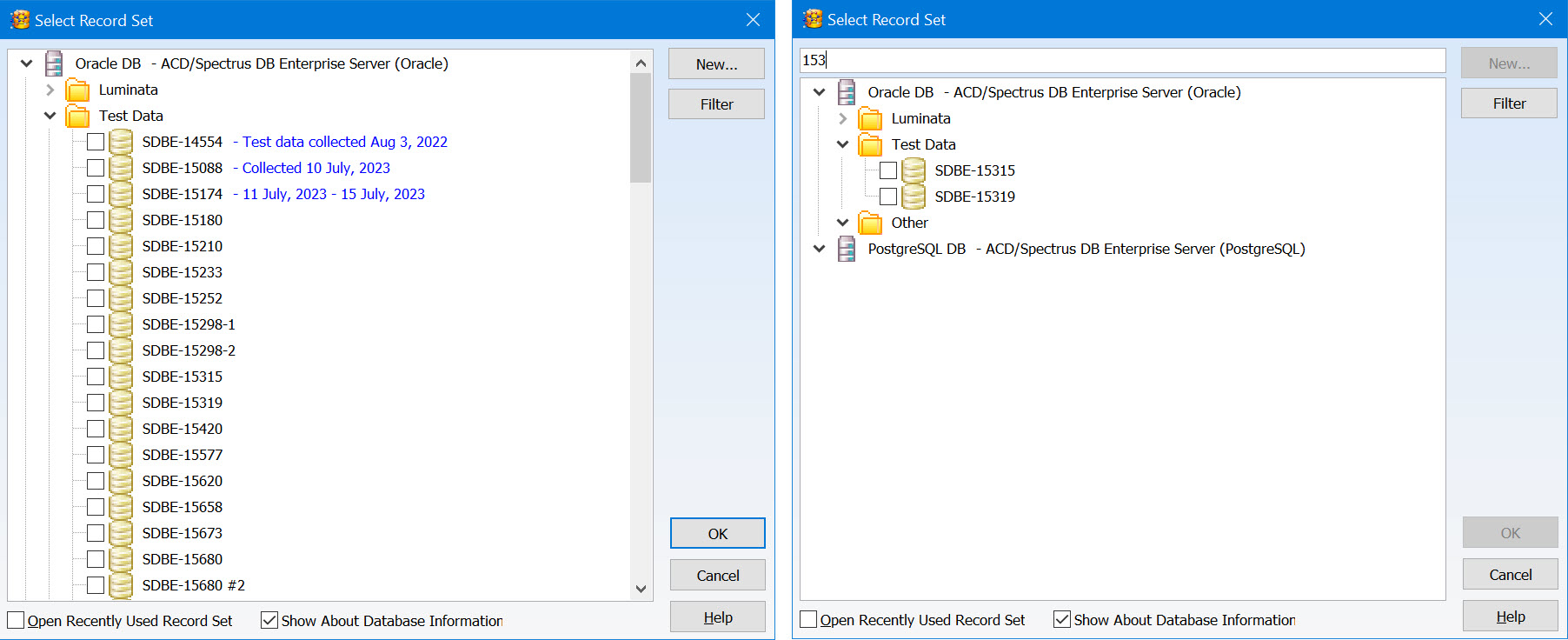
Enhanced Record Set Management
You now have additional features to help organize and review record sets, making it easier to find what you are looking for.
- Organize record sets into a tree layout using the Enterprise Server Management Console for greater control
- Add descriptive information to be displayed when you select a record set
- Text search is now available when reviewing record sets
Permission Management
Record owners can no longer open record sets by default. These permissions can be edited by a system administrator.
Expanded Database Search Options
You now have more tools for finding the data you need within database records.
- Search for records where a given table exists or not
- Common mass spectrometry related queries can now be done for a remote database
- Column names are now searchable for all tables in database records
- Perform further searches within the results of a previous search
Visualize Your Graph Data More Easily
- Review large data sets in the database more easily through faster display speed
As always, you can process data from other analytical techniques in Method Selection Suite. We’ve improved features for these techniques as well:
Processing MS & Chrom Data with Method Selection Suite
Improved Peak Naming for xC/UV/MS Data
Fine-tune peak naming for xC/UV/MS data with new options to:
- Automatically name your peak as a prefix + (optional) number suffix or use selected attributes (i.e., metadata, spectral parameters, etc.)
- Import peak names from your CDS
- Transfer peak names to the Table of Components and keep user-defined names
Improved Peak Detection and Integration for Hyphenated Data
You now have new options for the detection and integration of peaks:
- Specify an m/z range for spectral searching allowing the exclusion of extraneous ions and improving HQI (hit quality index) score
- Set a minimum area (% of max) threshold
- Combine adjacent and overlapping peaks by maximum resolution, and maximum number of peaks, and apply them to a specified region
- Apply minimum and maximum FWHM thresholds to flat chromatogram traces
- Equalize liftoff and touchdown heights
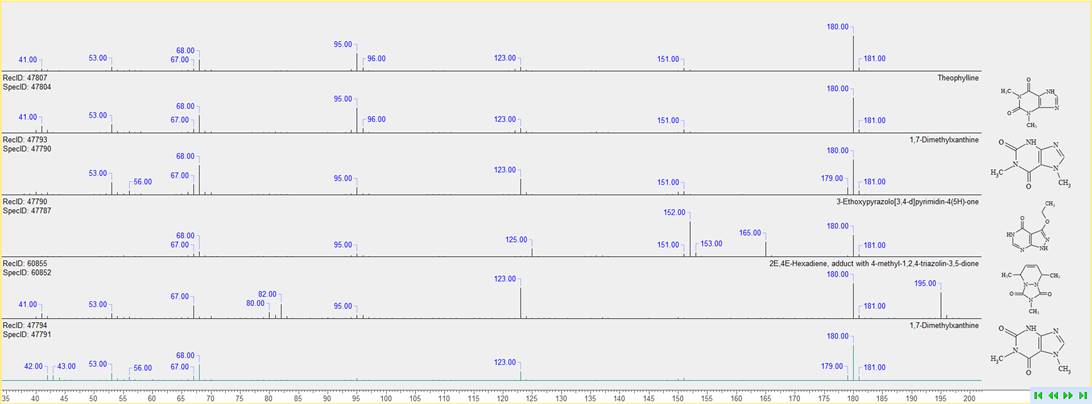
Consolidated Display of Results from Mass Spectral Search
You can now review mass spectral search results more conveniently and find the correct match through concurrent display of the query spectrum with hit spectra, structures, and metadata.
Account for Blank Injections for Sample Cleanup in LC/MS Data
You can now subtract flat chromatogram curves from one another to account for blank injections.
Ease of Use
Set method details (i.e., gradient, flow rate, etc.,) once and apply across all traces
Processing NMR Data with Method Selection Suite
Include Integral Assessment in Structure Verification
You can now assess how well the experimental 1H integral values match those predicted for the complete structure using the integral maximum and standard deviations in the Structure User Data table.
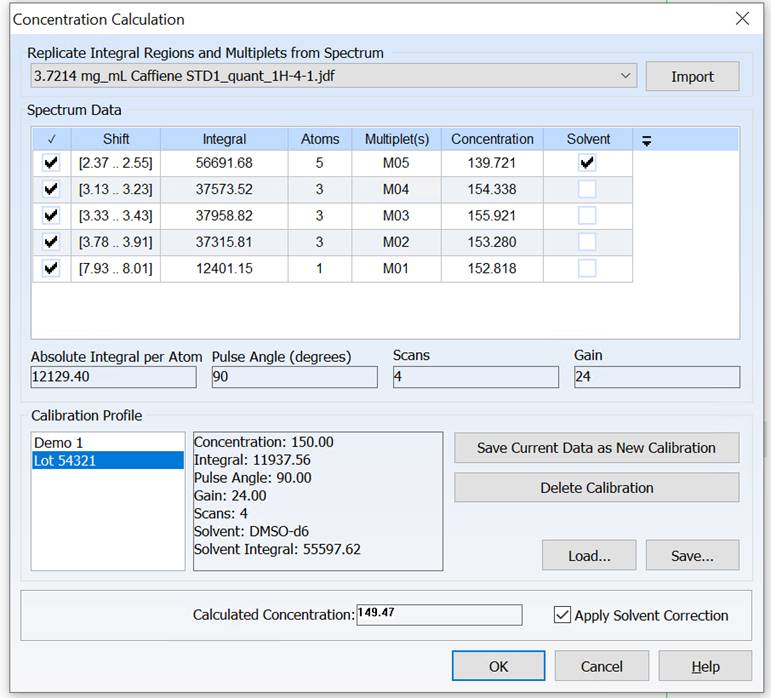
Adjust Solvent Integral to Minimize Error in External Standard qNMR
You can now compensate for error introduced by variability between NMR tubes when using an external standard for qNMR by normalizing the solvent integral in your spectrum with respect to that of the calibration spectrum.
The below features were included in the software in Spring 2023.
New Features for MS & Chrom Data Processing
Inclusion of NIST MS Search
- You can now use the NIST MS Search in spectral searching (also available as part of the IXCR workflow)

Obtain component name, MF (match factor), RMF (relative match factor) and probability of spectral match directly from NIST
Improved Hit Refinement for GC/MS Data with Component Deconvolution Workflows
- The software now supports radical cation (+.) structures. You can now automatically identify radical cations in data interpretation.
Improved Features for MS & Chrom Data Processing
Greater flexibility when working with series of flat chromatograms in hyphenated data sets (LC/UV/MS, GC/MS)
- Flat chromatograms and hyphenated data can be combined as series and saved as separate *.spectrus files. Table of Components, structures and biotransformation maps can be saved for each trace.

xC/UV/MS files can be collected as a series, saved as separate *.spectrus files and contain tables linked to each trace.
Transfer Peaks to the Table of Components
- You can now choose to merge or overwrite when transferring named peaks to the Table of Components
Filter Spectral Search Results by Spectrum Polarity
- You can now choose to filter spectral search results by spectrum polarity to ensure hits are relevant to your data
Improvements in Processing Multiply Charged Ions
Improved Verification of Multiply Charged Ions
- Now you can include multiply charged ions in the MS match verification of your molecular formula or structure
Find Target Ions
- You can now find target ions when any charge state (+/- 6) is present.
Set Filters for Auto Peak Picking
- You can now set a minimum USP signal-to-noise ratio to filter out peaks selected in the automatic peak picking algorithm
Report MSn Spectra
- You can choose to report multiple spectra by collision energy range and/or a specified value(s)
Increased Flexibility in Reporting LC/MS Data
- You can report peaks and components, one-per-page
Ease of Use Improvements
- Set the number of decimal places for mass values rounding before calculating of Mass Error
- Set the number of decimal places to be displayed for peak retention time and area values
- Isotope clusters can now be highlighted in the spectrum for formulae generated by Formula Generator and placed in the Table of Formulae
- There is now a new keyboard shortcut for horizontal zooming in spectra and chromatograms
New Features for NMR Data Processing
Process NUS Data with New Fast Algorithm
- You can now reconstruct 2D raw data acquired in Non-Uniform-Sampling (NUS) mode with a new, faster algorithm
Reference X Nuclei Spectra by Absolute Frequency
- You can now perform absolute referencing of the X nucleus (e.g., 15N, 19F, 31P, etc.) axis of 1D and 2D spectra based on a starting 1H or 13C spectrum
- For Bruker/TopSpin spectra of the same sample recorded consecutively, you use the observed absolute frequency of the reference signal from the 1H or 13C 1D spectra to reference the X spectra
Identify Overlayed 2D Spectra from New Color Legend
- You can now see which color corresponds to which spectrum in a legend when overlaying 2D spectra

Legend shows which spectrum is assigned to each color when working with multiple overlayed 2D spectra.
Use *.spectrus Files in Group Macros
- You can now execute group macros on multiple *.spectrus files
Improved Features for NMR Data Processing
Better Handling of Multiplets on the Side of Broad Water Peaks
- You can now get a more accurate integration of multiplets found on the edges of broad water peaks by enabling the Ignore Broad Water Peak option in your multiplet analysis

Perform more accurate integrations of multiplets on the sides of a broad water peak.
New Fields in the Table of User Data
- You can now see the reference and signal that were used in the Table of User Data
- You can now see the sum of all relative integrals for 1H spectra in the Table of User Data
See When Fourier Transform is Active in Preprocessing
- You can now see when Fourier transform is active in the Preprocessing Options to prevent disruptions to subsequent tasks
Apply 1D and 2D Preferences to all Open 1D and 2D Spectra
- You can now apply a set of preferences to all open 1D or 2D spectra with a single mouse-click, instead of having to repeat for each spectrum
Set Filters for Auto-Peak Picking
- You can now set a minimum USP signal to noise ratio to filter out peaks selected in the automatic peak-picking algorithm